A tour through HTSeq¶
In the analysis of high-throughput sequencing data, it is often necessary to write custom scripts to form the “glue” between tools or to perform specific analysis tasks. HTSeq is a Python package to facilitate this.
This tour demonstrates the functionality of HTSeq by performing a number of common analysis tasks:
- Getting statistical summaries about the base-call quality scores to study the data quality.
- Calculating a coverage vector and exporting it for visualization in a genome browser.
- Reading in annotation data from a GFF file.
- Assigning aligned reads from an RNA-Seq experiments to exons and genes.
The following description assumes that the reader is familiar with Python and with HTS data. (For a good and not too lengthy introduction to Python, read the Python Tutorial on the Python web site.)
If you want to try out the examples on your own system, you can download the example files used from here: HTSeq_example_data.tgz
Reading in reads¶
In the example data, a FASTQ file is provided with example reads from a yeast RNA-Seq
experiment. The file yeast_RNASeq_excerpt_sequence.txt is an excerpt of the
_sequence.txt file produced by the SolexaPipeline software. We can access it from
HTSeq with
>>> import HTSeq
>>> fastq_file = HTSeq.FastqReader( "yeast_RNASeq_excerpt_sequence.txt", "solexa" )
The first argument is the file name. The optional second argument indicates the encoding for the quality string. If you omit, the default (“phred”) is used. The example data, however, is from an older experiment, and hence encoded in the offset-64 format that the Solexa/Illumina software pipeline used before version 1.8. (A third option is “solexa_old”, for data from the Solexa pipeline prior to version 1.3.)
The variable fastq_file is now an object of class FastqReader, which
refers to the file:
>>> fastq_file
<FastqReader object, connected to file name 'yeast_RNASeq_excerpt_sequence.txt'>
When used in a for loop, it generates an iterator of objects representing the
reads. Here, we use the islice function from itertools to cut after 10
reads.
>>> import itertools
>>> for read in itertools.islice( fastq_file, 10 ):
... print read
CTTACGTTTTCTGTATCAATACTCGATTTATCATCT
AATTGGTTTCCCCGCCGAGACCGTACACTACCAGCC
TTTGGACTTGATTGTTGACGCTATCAAGGCTGCTGG
ATCTCATATACAATGTCTATCCCAGAAACTCAAAAA
AAAGTTCGAATTAGGCCGTCAACCAGCCAACACCAA
GGAGCAAATTGCCAACAAGGAAAGGCAATATAACGA
AGACAAGCTGCTGCTTCTGTTGTTCCATCTGCTTCC
AAGAGGTTTGAGATCTTTGACCACCGTCTGGGCTGA
GTCATCACTATCAGAGAAGGTAGAACATTGGAAGAT
ACTTTTAAAGATTGGCCAAGAATTGGGGATTGAAGA
Of course, there is more to a read than its sequence. The variable read still
contains the tenth read, and we can examine it:
>>> read
<SequenceWithQualities object 'HWI-EAS225:1:10:1284:142#0/1'>
A Sequence object has two slots, called seq and
name. This object is a SequenceWithQualities,
and it also has a slot qual:
>>> read.name
'HWI-EAS225:1:10:1284:142#0/1'
>>> read.seq
'ACTTTTAAAGATTGGCCAAGAATTGGGGATTGAAGA'
>>> read.qual
array([33, 33, 33, 33, 33, 33, 29, 27, 29, 32, 29, 30, 30, 21, 22, 25, 25,
25, 23, 28, 24, 24, 29, 29, 29, 25, 28, 24, 24, 26, 25, 25, 24, 24,
24, 24])
The values in the quality array are, for each base in the sequence, the Phred score for the correctness of the base.
As a first simple example for the use of HTSeq, we now calculate the average
quality score for each position in the reads by adding up the qual arrays
from all reads and the dividing by the number of reads. We sum everything up in
the variable qualsum, a numpy array of integers:
>>> import numpy
>>> len( read )
36
>>> qualsum = numpy.zeros( len(read), numpy.int )
Then we loop through the fastq file, adding up the quality scores and counting the reads:
>>> nreads = 0
>>> for read in fastq_file:
... qualsum += read.qual
... nreads += 1
The average qualities are hence:
>>> qualsum / float(nreads)
array([ 31.56838274, 30.08288332, 29.4375375 , 29.00432017,
28.55290212, 28.26825073, 28.46681867, 27.59082363,
27.34097364, 27.57330293, 27.11784471, 27.19432777,
26.84023361, 26.76267051, 26.44885795, 26.79135165,
26.42901716, 26.49849994, 26.13604544, 25.95823833,
25.54922197, 26.20460818, 25.42333693, 25.72298892,
25.04164167, 24.75151006, 24.48561942, 24.27061082,
24.10720429, 23.68026721, 23.52034081, 23.49437978,
23.11076443, 22.5576223 , 22.43549742, 22.62354494])
If you have matplotlib installed, you can plot these numbers.
>>> from matplotlib import pyplot
>>> pyplot.plot( qualsum / nreads )
[<matplotlib.lines.Line2D object at 0x...>]
>>> pyplot.show()
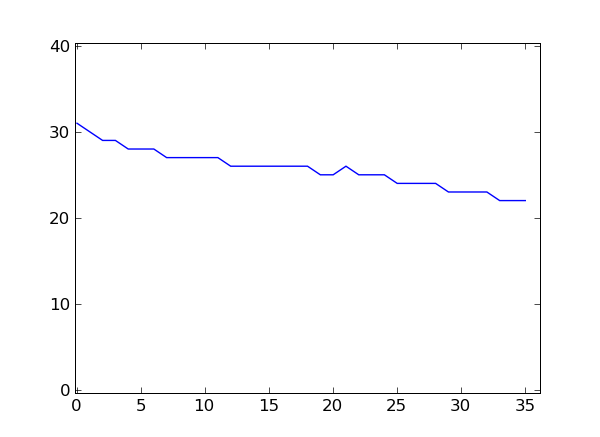
This is a very simple way of looking at the quality scores. For more sophisticated quality-control techniques, see the Chapter Quality Assessment with htseq-qa.
Instead of a FASTQ file, you might have a SAM file, with the reads already aligned. The SAM_Reader class can read such data.
>>> alignment_file = HTSeq.SAM_Reader( "yeast_RNASeq_excerpt.sam" )
If we are only interested in the qualities, we can rewrite the commands from above
to use the alignment_file:
>>> nreads = 0
>>> for aln in alignment_file:
... qualsum += aln.read.qual
... nreads += 1
We have simple replaced the FastqReader with a SolexaExportReader, which
iterates, when used in a for loop, over SolexaExportAlignment objects. Each of
these contain a field read that contains the SequenceWithQualities
object, as before. There are more parses, for example the SAM_Reader that can read SAM
files, and generates SAM_Alignment objects. As all Alignment objects
contain a read slot with the SequenceWithQualities, we can use the same
code with any alignment file for which a parser has been provided, and all we have
to change is the name of the reader class in the first line.
The other fields that all Alignment objects contain, is a Boolean called
aligned that tells us whether the read has been aligned
at all, and a field called iv
(for “interval”) that shows where the read was aligned to. We use this information in
the next section.
Reading and writing BAM files¶
HTSeq exposes the samtools API trough pysam, enabling you to read and write BAM files. A simple example of the usage is given here:
>>> bam_reader = HTSeq.BAM_Reader( "SRR001432_head_sorted.bam" )
>>> for a in itertools.islice( bam_reader, 5 ): # printing first 5 reads
... print a
<SAM_Alignment object: Read 'SRR001432.165255 USI-EAS21_0008_3445:8:4:718:439 length=25' aligned to 1:[29267,29292)/->
<SAM_Alignment object: Read 'SRR001432.238475 USI-EAS21_0008_3445:8:6:888:446 length=25' aligned to 1:[62943,62968)/->
<SAM_Alignment object: Read 'SRR001432.116075 USI-EAS21_0008_3445:8:3:657:64 length=25' aligned to 1:[86980,87005)/->
<SAM_Alignment object: Read 'SRR001432.159692 USI-EAS21_0008_3445:8:4:618:821 length=25' aligned to 1:[91360,91385)/->
<SAM_Alignment object: Read 'SRR001432.249247 USI-EAS21_0008_3445:8:6:144:741 length=25' aligned to 1:[97059,97084)/->
[FIXME] The following is currently broken, likely due to a bug in pysam.
>>> bam_writer = HTSeq.BAM_Writer.from_BAM_Reader( "region.bam", bam_reader ) #set-up BAM_Writer with same header as reader
>>> for a in bam_reader.fetch( region = "1:249000000-249200000" ): #fetching reads in a region
... print "Writing Alignment", a, "to file", bam_writer.filename
... bam_writer.write( a )
>>> bam_writer.close()
Genomic intervals and genomic arrays¶
Genomic intervals¶
At the end of the previous section, we looped through a SAM file. In the for loop,
the SAM_Reader object yields for each alignment line in the SAM file an
object of class SAM_Alignment. Let’s have closer look at such an object,
still found in the variable aln:
>>> aln
<SAM_Alignment object: Read 'HWI-EAS225:1:11:76:63#0/1' aligned to IV:[246048,246084)/+>
Every alignment object has a slot read, that contains a SequenceWithQualities object as
described above
>>> aln.read
<SequenceWithQualities object 'HWI-EAS225:1:11:76:63#0/1'>
>>> aln.read.name
'HWI-EAS225:1:11:76:63#0/1'
>>> aln.read.seq
'ACTGTAAATACTTTTCAGAAGAGATTTGTAGAATCC'
>>> aln.read.qual
array([33, 33, 33, 33, 31, 33, 30, 32, 33, 30, 29, 33, 32, 32, 32, 31, 32,
31, 29, 28, 30, 28, 30, 24, 28, 30, 28, 26, 24, 29, 24, 23, 23, 27,
25, 25])
Furthermore, every alignment object has a slot iv (for “interval”) that describes where
the read was aligned to (if it was aligned). To hold this
information, an object of class GenomicInterval
is used that has slots as follows:
>>> aln.iv
<GenomicInterval object 'IV', [246048,246084), strand '+'>
>>> aln.iv.chrom
'IV'
>>> aln.iv.start
246048
>>> aln.iv.end
246084
>>> aln.iv.strand
'+'
Note that all coordinates in HTSeq are zero-based (following Python convention), i.e.
the first base of a chromosome has index 0. Also, all intervals are half-open, i.e.,
the end position is not included. The strand can be one of '+', '-',
and '.', where the latter indicates that the strand is not defined or not of interest.
Apart from these slots,
a GenomicInterval object has a number of convenience functions, see the reference.
Note that a SAM file may contain reads that could not be aligned. For these, the iv slot contains None. To test whether an alignment is present, you can also query the slot aligned, which is a Boolean.
Genomic Arrays¶
The GenomicArray data structure is a convenient way to store and
retrieve information associated with a genomic position or genomic interval. In
a GenomicArray, data (either simple scalar data like a number) or can be stored
at a place identified by a GenomicInterval. We demonstrate with a toy example.
Assume you have a genome with three chromosomes with the following lengths (in bp):
>>> chromlens = { 'chr1': 3000, 'chr2': 2000, 'chr1': 1000 }
We wish to store integer data (typecode “i”)
>>> ga = HTSeq.GenomicArray( chromlens, stranded=False, typecode="i" )
Now, we can assign the value 5 to an interval:
>>> iv = HTSeq.GenomicInterval( "chr1", 100, 120, "." )
>>> ga[iv] = 5
We may want to add the value 3 to an interval overlapping with the previous one:
>>> iv = HTSeq.GenomicInterval( "chr1", 110, 135, "." )
>>> ga[iv] += 3
To see the effect of this, we read out an interval encompassing the region that we changed. To display the data, we convert to a list:
>>> iv = HTSeq.GenomicInterval( "chr1", 90, 140, "." )
>>> list( ga[iv] )
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 5, 5, 5, 5, 5, 5, 5, 5, 5, 5, 8, 8, 8,
8, 8, 8, 8, 8, 8, 8, 3, 3, 3, 3, 3, 3, 3, 3, 3, 3, 3, 3, 3, 3, 3, 0,
0, 0, 0, 0]
It would be wasteful to store all these repeats of the same value as it is displayed here. Hence, GenomicArray objects use by default so-called StepVectors that store the data internally in “steps” of constant value. Often, reading out the data that way is useful, too:
>>> for iv2, value in ga[iv].steps():
... print iv2, value
...
chr1:[90,100)/. 0
chr1:[100,110)/. 5
chr1:[110,120)/. 8
chr1:[120,135)/. 3
chr1:[135,140)/. 0
If the steps become very small, storing them instead of just the unrolled data may
become inefficient. In this case, GenomicArrays should be instantiated with
storage mode ndarray to get a normal numpy array as backend, or with storage
mode memmap to use a file/memory-mapped numpy array (see reference for details).
In the following section, we demonstrate how a GenomicArray can be used to calculate a coverage vector. In the section after that, we see how a GenomicArray with type code ‘O’ (which stands for ‘object’, i.e., any kind of data, not just numbers) is useful to organize metadata.
Calculating coverage vectors¶
By a “coverage vector”, we mean a vector (one-dimensional array) of the length of a chromosome, where each element counts how many reads cover the corresponding base pair in their alignment. A GenomicArray can conveniently bundle the coverage vectors for all the chromosomes in a genome.
Hence, we start by defining a GenomicArray:
>>> cvg = HTSeq.GenomicArray( "auto", stranded=True, typecode="i" )
Instead of listing all chromosomes, we instruct the GenomicArray to add chromosome
vectors as needed, by specifiyng "auto". As we set stranded=True, there are now two
chromosome vectors for each
chromosome, all holding integer values (typecode="i"). They all have an
“infinte” length as we did not specify the actual lengths of the chromosomes.
To build the coverage vectors, we now simply iterate through all the reads and add the value 1 at the interval to which each read was aligned to:
>>> alignment_file = HTSeq.SAM_Reader( "yeast_RNASeq_excerpt.sam" )
>>> cvg = HTSeq.GenomicArray( "auto", stranded=True, typecode='i' )
>>> for alngt in alignment_file:
... if alngt.aligned:
... cvg[ alngt.iv ] += 1
We can plot an excerpt of this with:
>>> pyplot.plot( list( cvg[ HTSeq.GenomicInterval( "III", 200000, 500000, "+" ) ] ) )
[<matplotlib.lines.Line2D object at 0x...>]
However, a proper genome browser gives a better impression of the data. The following commands write two BedGraph (Wiggle) files, one for the plus and one for the minus strands:
>>> cvg.write_bedgraph_file( "plus.wig", "+" )
>>> cvg.write_bedgraph_file( "minus.wig", "-" )
These two files can then be viewed in a genome browser (e.g. IGB or IGV), alongside the annotation from a GFF file (see below).
GenomicArrayOfSets¶
Another use of genomic arrays is to store annotation data. In the next section, we will use this to store the position of all exons of the yeast genome in a genomic array and then go through all our reads, querying the array for each read to report the exons overlapped by this read.
In principle, we could use a genomic array with type code ‘O’ (for object), which can store arbitrary Python objects. However, there might be positions in the genome that are covered by more than one gene, and hence, we better use a data structure that can accommodate overlapping features.
The class:GenomicArrayOfSets is meant for this purpose. For each step, it stores a set of objects. To
illustrate this, we initialize a GenomicArrayOfSets and then store three features in it:
>>> gas = HTSeq.GenomicArrayOfSets( "auto", stranded=False )
>>> gas[ HTSeq.GenomicInterval( "chr1", 100, 250 ) ] += "A"
>>> gas[ HTSeq.GenomicInterval( "chr1", 360, 640 ) ] += "A"
>>> gas[ HTSeq.GenomicInterval( "chr1", 510, 950 ) ] += "B"
These three features represent three exons of two genes, arranged as shown in this figure:

Note that we used +=, not just =, above when adding the features. With a GenomicArrayOfSets, you need to always use the += operator (rather than =), so that the values gets added to the step’s set.
Now consider a read that aligns to the following interval (represented in the figure above by the light blue line ):
>>> read_iv = HTSeq.GenomicInterval( "chr1", 450, 800 )
We can query the GenomicArrayOfSets, as before:
>>> for iv, val in gas[ read_iv ].steps():
... print iv, val
chr1:[450,510)/. set(['A'])
chr1:[510,640)/. set(['A', 'B'])
chr1:[640,800)/. set(['B'])
The interval has been subdivided into three pieces, corresponding to the three different sets that it overlaps, namely first only A, then A and B, and finally only B.
You might be only interested in the set of all features that the read interval overlaps. To this end, just
form the set union of the three reported sets, using Python’s set union operator (|):
>>> fset = set()
>>> for iv, val in gas[ read_iv ].steps():
... fset |= val
>>> print fset
set(['A', 'B'])
Experienced Python developers will recognize that the for loop can be replaced with a single line
using a generator comprehension and the reduce function:
>>> reduce( set.union, ( val for iv, val in gas[ read_iv ].steps() ) )
set(['A', 'B'])
We will come back to the constructs in the next section, after a brief detour on how to read GTF files.
Counting reads by genes¶
As the example data is from an RNA-Seq experiment, we want to know how many reads fall into the exonic regions of each gene. For this purpose we first need to read in information about the positions of the exons. A convenient source of such information are the GTF files from Ensembl (to be found here).
These file are in the GTF format, a tightening of the GFF format. HTSeq offers the
GFF_Reader class to read in a GFF file:
>>> gtf_file = HTSeq.GFF_Reader( "Saccharomyces_cerevisiae.SGD1.01.56.gtf.gz",
... end_included=True )
The GFF format is, unfortunately, a not very well specified file format. Several standard documents exist, from different groups, which contradict each other in some points. Most importantly, it is unclear whether a range specified in a GFF line is supposed to include the base under the “end” position or not. Here, we specied the this file does include the end. Actually, this is the default for GFF_Reader, so it would not have been necessary to specify it. (Hint, if you are unsure about your GFF file: The length of most coding exons is divisible by 3. If start-end is divisible by 3, too, end is not included, if the division leaves a remainder of two, end is included.)
We iterate through this file as follows:
>>> for feature in itertools.islice( gtf_file, 10 ):
... print feature
...
<GenomicFeature: exon 'R0010W' at 2-micron: 251 -> 1523 (strand '+')>
<GenomicFeature: CDS 'R0010W' at 2-micron: 251 -> 1520 (strand '+')>
<GenomicFeature: start_codon 'R0010W' at 2-micron: 251 -> 254 (strand '+')>
<GenomicFeature: stop_codon 'R0010W' at 2-micron: 1520 -> 1523 (strand '+')>
<GenomicFeature: exon 'R0020C' at 2-micron: 3007 -> 1885 (strand '-')>
<GenomicFeature: CDS 'R0020C' at 2-micron: 3007 -> 1888 (strand '-')>
<GenomicFeature: start_codon 'R0020C' at 2-micron: 3007 -> 3004 (strand '-')>
<GenomicFeature: stop_codon 'R0020C' at 2-micron: 1888 -> 1885 (strand '-')>
<GenomicFeature: exon 'R0030W' at 2-micron: 3270 -> 3816 (strand '+')>
<GenomicFeature: CDS 'R0030W' at 2-micron: 3270 -> 3813 (strand '+')>
The feature variable is filled with objects of class GenomicFeature.
If you compare the coordinated with the original file, you will notice that the
GFF_Reader has subtracted one from all starts. This is because all file parsers in
HTSeq adjust coordinates as necessary to fit the Python convention, which is that
indexing starts with zero and the end is not included. Hence, you can immediately
compare coordinates from different data formats without having to worry about
subtleties like the fact that GFF is one-based and SAM is zero-based.
As with all Python objects, the dir function shows us the slots and
functions of our loop variable feature and so allow us to inspect what data
it provides:
>>> dir( feature )
['__class__', ..., '__weakref__', 'attr', 'frame', 'get_gff_line',
'iv', 'name', 'score', 'source', 'type']
Ignoring the attributes starting with an underscore, we can see now how to access the information stored in the GFF file. The information from the columns of the GFF table is accessible as follows:
>>> feature.iv
<GenomicInterval object '2-micron', [3270,3813), strand '+'>
>>> feature.source
'protein_coding'
>>> feature.type
'CDS'
>>> feature.score
'.'
The last column (the attributes) is parsed and presented as a dict:
>>> feature.attr
{'exon_number': '1', 'gene_id': 'R0030W', 'transcript_name': 'RAF1',
'transcript_id': 'R0030W', 'protein_id': 'R0030W', 'gene_name': 'RAF1'}
The very first attribute in this column is usually some kind of ID, hence it is
stored in the slot name:
>>> feature.name
'R0030W'
To deal with this data, we will use the GenomicArrayOfSets introuced in the
previous section.
>>> exons = HTSeq.GenomicArrayOfSets( "auto", stranded=False )
However, our RNA-Seq experiment was not strand-specific, i.e., we do not know whether
the reads came from the plus or the minus strand. This is why we defined the GenomicArrayOfSet
as non-stranded (stranded=False in the instantiation of exons above), instructing
it to ignore all strand information. Teherfore, we now have many overlapping
genes, but the GenomicArrayOfSets will handle this.
>>> for feature in gtf_file:
... if feature.type == "exon":
... exons[ feature.iv ] += feature.name
Nate that, we only store the gene name this time, as this will be more convenient later.
Assume we have a read covering this interval:
>>> iv = HTSeq.GenomicInterval( "III", 23850, 23950, "." )
Its left half covers two genes (YCL058C, YCL058W-A), but its right half only YCL058C because YCL058W-A end in the middle of the read:
>>> list( exons[iv].steps() )
[(<GenomicInterval object 'III', [23850,23925), strand '.'>,
set(['YCL058C', 'YCL058W-A'])),
(<GenomicInterval object 'III', [23925,23950), strand '.'>,
set(['YCL058C']))]
Assuming the transcription boundaries in our GTF file to be correct, we may conclude that this read is from the gene that appears in both steps and not from the one that appears in only one of the steps. More generally, whenever a read overlaps multiple steps (a new step starts wherever a feature starts or ends), we get a set of feature names for each step, and we have to find the intersection of all these. This can be coded as follows:
>>> iset = None
>>> for iv2, step_set in exons[iv].steps():
... if iset is None:
... iset = step_set.copy()
... else:
... iset.intersection_update( step_set )
...
>>> print iset
set(['YCL058C'])
When we look at the first step, we make a
copy of the steps (in order to not disturb the values stored in exons.) For the following
steps, we use the intersection_update
method Python’s standard set class, which performs a set intersection in
place. Afterwards, we have a set with precisely one element. Getting this one
element is a tiny bit cumbersome; to access it, one needs to write:
>>> list(iset)[0]
'YCL058C'
In this way, we can go through all our aligned reads, calculate the intersection set, and, if it contains a single gene name, add a count for this gene. For the counters, we use a dict, which we initialize with a zero for each gene name:
>>> counts = {}
>>> for feature in gtf_file:
... if feature.type == "exon":
... counts[ feature.name ] = 0
Now, we can finally count:
>>> sam_file = HTSeq.SAM_Reader( "yeast_RNASeq_excerpt.sam" )
>>> for alnmt in sam_file:
... if alnmt.aligned:
... iset = None
... for iv2, step_set in exons[ alnmt.iv ].steps():
... if iset is None:
... iset = step_set.copy()
... else:
... iset.intersection_update( step_set )
... if len( iset ) == 1:
... counts[ list(iset)[0] ] += 1
We can now conveniently print the result with:
>>> for name in sorted( counts.keys() ):
... print name, counts[name]
15S_rRNA 0
21S_rRNA 0
HRA1 0
...
YPR048W 2
YPR049C 3
YPR050C 0
YPR051W 1
YPR052C 1
YPR053C 5
YPR054W 0
...
tY(GUA)M2 0
tY(GUA)O 0
tY(GUA)Q 0
Some aligners can output gapped or spliced alignments. In a SAM file, this in encoded
in the CIGAR string. HTSeq has facilities to handle this conveniently, too, with the
class CigarOperation. Chapter Counting reads in features with htseq-count describes a script which offers
some further counting schemes.
And much more¶
This tour is only meant to give an overview. There are many more tasks that can be solved with HTSeq. Have a look at the reference documentation in the following pages to see what else is there.