File input and output¶
See also
The ase.io module has three basic functions: read(),
iread() and write(). The methods are described here:
- ase.io.read(filename: Union[str, PurePath, IO], index: Any = None, format: str = None, parallel: bool = True, do_not_split_by_at_sign: bool = False, **kwargs) Union[Atoms, List[Atoms]][source]¶
Read Atoms object(s) from file.
- filename: str or file
Name of the file to read from or a file descriptor.
- index: int, slice or str
The last configuration will be returned by default. Examples:
index=0: first configurationindex=-2: second to lastindex=':'orindex=slice(None): allindex='-3:'orindex=slice(-3, None): three lastindex='::2'orindex=slice(0, None, 2): evenindex='1::2'orindex=slice(1, None, 2): odd
- format: str
Used to specify the file-format. If not given, the file-format will be guessed by the filetype function.
- parallel: bool
Default is to read on master and broadcast to slaves. Use parallel=False to read on all slaves.
- do_not_split_by_at_sign: bool
If False (default)
filenameis splited by at sign@
Many formats allow on open file-like object to be passed instead of
filename. In this case the format cannot be auto-decected, so theformatargument should be explicitly given.
- ase.io.iread(filename: Union[str, PurePath, IO], index: Any = None, format: str = None, parallel: bool = True, do_not_split_by_at_sign: bool = False, **kwargs) Iterable[Atoms][source]¶
Iterator for reading Atoms objects from file.
Works as the \(read\) function, but yields one Atoms object at a time instead of all at once.
- ase.io.write(filename: Union[str, PurePath, IO], images: Union[Atoms, Sequence[Atoms]], format: str = None, parallel: bool = True, append: bool = False, **kwargs: dict) None[source]¶
Write Atoms object(s) to file.
- filename: str or file
Name of the file to write to or a file descriptor. The name ‘-’ means standard output.
- images: Atoms object or list of Atoms objects
A single Atoms object or a list of Atoms objects.
- format: str
Used to specify the file-format. If not given, the file-format will be taken from suffix of the filename.
- parallel: bool
Default is to write on master only. Use parallel=False to write from all slaves.
- append: bool
Default is to open files in ‘w’ or ‘wb’ mode, overwriting existing files. In some cases opening the file in ‘a’ or ‘ab’ mode (appending) is useful, e.g. writing trajectories or saving multiple Atoms objects in one file. WARNING: If the file format does not support multiple entries without additional keywords/headers, files created using ‘append=True’ might not be readable by any program! They will nevertheless be written without error message.
The use of additional keywords is format specific. write() may return an object after writing certain formats, but this behaviour may change in the future.
Use ase info --formats to see a list of formats. This information
is programmatically accessible as ase.io.formats.ioformats, a
dictionary which maps format names to ase.io.formats.IOFormat
objects.
These are the file-formats that are recognized (formats with a + support
multiple configurations):
format |
description |
capabilities |
|---|---|---|
ABINIT input file |
RW |
|
ABINIT output file |
R |
|
ACE input file |
R |
|
ACE output file |
R |
|
FHI-aims geometry file |
RW |
|
FHI-aims output |
R+ |
|
ASE bundle trajectory |
RW+ |
|
CASTEP output file |
R+ |
|
CASTEP geom file |
RW |
|
CASTEP trajectory file |
R+ |
|
CASTEP molecular dynamics file |
R+ |
|
CASTEP phonon file |
R |
|
AtomEye configuration |
RW |
|
CIF-file |
RW+ |
|
CMDFT-file |
R |
|
Chemical json file |
R |
|
CP2K DCD file |
R+ |
|
CP2K restart file |
R |
|
Crystal fort.34 format |
RW |
|
CUBE file |
RW |
|
Dacapo text output |
R |
|
ASE SQLite database file |
RW+ |
|
DftbPlus input file |
RW |
|
DL_POLY HISTORY file |
R+ |
|
DL_POLY_4 CONFIG file |
RW |
|
DMol3 arc file |
RW+ |
|
DMol3 structure file |
RW |
|
DMol3 structure file |
RW |
|
ELK atoms definition from GEOMETRY.OUT |
R |
|
ELK input file |
W |
|
EON CON file |
RW+ |
|
Encapsulated Postscript |
W |
|
Quantum espresso in file |
RW |
|
Quantum espresso out file |
R+ |
|
exciting input |
RW |
|
Extended XYZ file |
RW+ |
|
FINDSYM-format |
W+ |
|
GAMESS-US input file |
W |
|
GAMESS-US output file |
R |
|
GAMESS-US punchcard file |
R |
|
Gaussian com (input) file |
RW |
|
Gaussian output file |
R+ |
|
DFTBPlus GEN format |
RW |
|
Graphics interchange format |
W+ |
|
GPAW text output |
R+ |
|
GPUMD input file |
RW |
|
GPAW restart-file |
R |
|
Gromacs coordinates |
RW |
|
Gromos96 geometry file |
RW |
|
X3DOM HTML |
W |
|
ASE JSON database file |
RW+ |
|
JSV file format |
RW |
|
LAMMPS data file |
RW |
|
LAMMPS binary dump file |
R+ |
|
LAMMPS text dump file |
R+ |
|
MAGRES ab initio NMR data file |
RW |
|
MDL Molfile |
R |
|
MP4 animation |
W+ |
|
muSTEM xtl file |
RW |
|
ASE MySQL database file |
RW+ |
|
AMBER NetCDF trajectory file |
RW+ |
|
JSON from Nomad archive |
R+ |
|
NWChem input file |
RW |
|
NWChem output file |
R+ |
|
Octopus input file |
R |
|
Portable Network Graphics |
W |
|
ASE PostgreSQL database file |
RW+ |
|
Persistance of Vision |
W |
|
prismatic and computem XYZ-file |
RW |
|
Protein Data Bank |
RW+ |
|
Python file |
W+ |
|
QBOX output file |
R+ |
|
SHELX format |
RW |
|
RMCProfile |
RW |
|
SDF format |
R |
|
Siesta .XV file |
R |
|
WIEN2k structure file |
RW |
|
SIESTA STRUCT file |
R |
|
qball sys file |
RW |
|
ASE trajectory |
RW+ |
|
TURBOMOLE coord file |
RW |
|
TURBOMOLE gradient file |
R+ |
|
V_Sim ascii file |
RW |
|
VASP POSCAR/CONTCAR |
RW |
|
VASP OUTCAR file |
R+ |
|
VASP XDATCAR file |
RW+ |
|
VASP vasprun.xml file |
R+ |
|
VTK XML Image Data |
W |
|
VTK XML Unstructured Grid |
W |
|
Wannier90 output |
R |
|
X3D |
W |
|
Materials Studio file |
RW |
|
XCrySDen Structure File |
RW+ |
|
Materials Studio file |
RW+ |
|
XYZ-file |
RW+ |
Note
Even though that ASE does a good job reading the above listed formats, it may not read some unusual features or strangely formatted files.
For the CIF format, STAR extensions as save frames, global blocks, nested loops and multi-data values are not supported.
Note
ASE read and write functions are automatically parallelized if a
suitable MPI library is found. This requires to call read and write
with same input on all cores. For more information, see
ase.parallel.
Note
ASE can read and write directly to compressed files. Simply add .gz,
.bz2 or .xz to your filename.
The read() function is only designed to retrieve the atomic configuration
from a file, but for the CUBE format you can import the function:
- ase.io.read_cube_data()¶
which will return a (data, atoms) tuple:
from ase.io.cube import read_cube_data
data, atoms = read_cube_data('abc.cube')
Examples¶
>>> from ase import Atoms
>>> from ase.build import fcc111, add_adsorbate, bulk
>>> from ase.io import read, write
>>> adsorbate = Atoms('CO')
>>> adsorbate[1].z = 1.1
>>> a = 3.61
>>> slab = fcc111('Cu', (2, 2, 3), a=a, vacuum=7.0)
>>> add_adsorbate(slab, adsorbate, 1.8, 'ontop')
Write PNG image
>>> write('slab.png', slab * (3, 3, 1), rotation='10z,-80x')
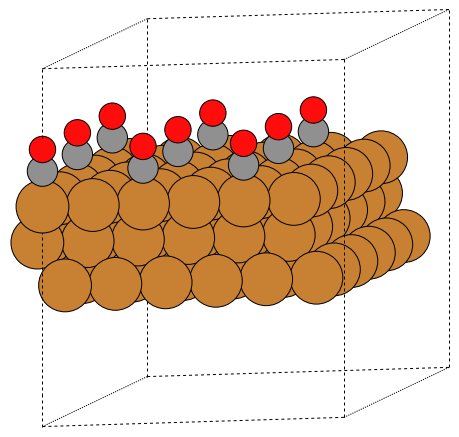
Write animation with 500 ms duration per frame
>>> write('movie.gif', [bulk(s) for s in ['Cu', 'Ag', 'Au']], interval=500)
Write POVRAY file (the projection settings and povray specific settings are separated)
>>> write('slab.pov', slab * (3, 3, 1),
... generic_projection_settings = dict(rotation='10z,-80x'))
This will write both a slab.pov and a slab.ini file. Convert
to PNG with the command povray slab.ini or use the
.render method on the returned object:
Here is an example using bbox
>>> d = a / 2**0.5
>>> write('slab.pov', slab * (2, 2, 1),
... generic_projection_settings = dict(
... bbox=(d, 0, 3 * d, d * 3**0.5))).render()

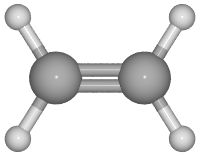
This is an example of displaying bond order for a molecule
# creates: C2H4.png
from ase.build.molecule import molecule
from ase.io import write
from ase.io.pov import get_bondpairs, set_high_bondorder_pairs
C2H4 = molecule('C2H4')
r = [{'C': 0.4, 'H': 0.2}[at.symbol] for at in C2H4]
bondpairs = get_bondpairs(C2H4, radius=1.1)
high_bondorder_pairs = {}
# This defines offset, bond order, and bond_offset of the bond between 0 and 1
high_bondorder_pairs[(0, 1)] = ((0, 0, 0), 2, (0.17, 0.17, 0))
bondpairs = set_high_bondorder_pairs(bondpairs, high_bondorder_pairs)
renderer = write('C2H4.pov', C2H4, format='pov',
radii=r, rotation='90y',
povray_settings=dict(canvas_width=200, bondatoms=bondpairs))
renderer.render()
Note that in general the XYZ-format does not contain information about the unit cell, however, ASE uses the extended XYZ-format which stores the unitcell:
>>> from ase.io import read, write
>>> write('slab.xyz', slab)
>>> a = read('slab.xyz')
>>> cell = a.get_cell()
>>> cell.round(3)
array([[ 5.105, 0. , 0. ],
[ 2.553, 4.421, 0. ],
[ 0. , 0. , 18.168]])
>>> a.get_pbc()
array([ True, True, False], dtype=bool)
Another way to include the unit cell is to write the cell vectors at the end of the file as VEC<N> <x> <y> <z> (used for example in the ADF software).
>>> write('slab.xyz', vec_cell=True)
Use ASE’s native format for writing all information:
>>> write('slab.traj', slab)
>>> b = read('slab.traj')
>>> b.cell.round(3)
array([[ 5.105, 0. , 0. ],
[ 2.553, 4.421, 0. ],
[ 0. , 0. , 18.168]])
>>> b.pbc
array([ True, True, False], dtype=bool)
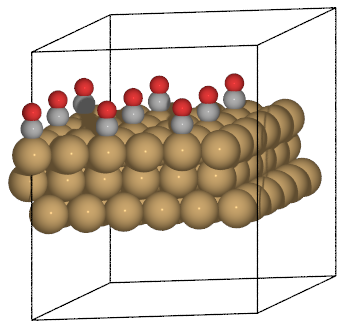
A script showing all of the povray parameters, and generating the image below,
can be found here: save_pov.py
An other example showing how to change colors and textures in pov can
be found here: ../../tutorials/saving_graphics.py.
Adding a new file-format to ASE¶
Try to model the read/write functions after the xyz format as implemented in :git:`ase/io/xyz.py` and also read, understand and update :git:`ase/io/formats.py`.